
API Usage Examples#
HBAT 2 provides interactive Jupyter notebooks demonstrating various features and use cases with 3D molecular visualizations.
Example Notebooks#
The following notebooks are available in the notebooks/ directory:
Notebook |
Description |
Open in Colab |
|---|---|---|
Comprehensive analysis of 6RSA (Ribonuclease A) structure including hydrogen bonds, cooperativity chains, and 3D visualization with py3Dmol |
|
|
Halogen bond detection and visualization in 4X21 crystal structure, demonstrating C-X···A interactions with interactive 3D views |
|
|
Comparing PDBFixer vs OpenBabel for hydrogen addition, analyzing determinism and hydrogen bond count variations |
|
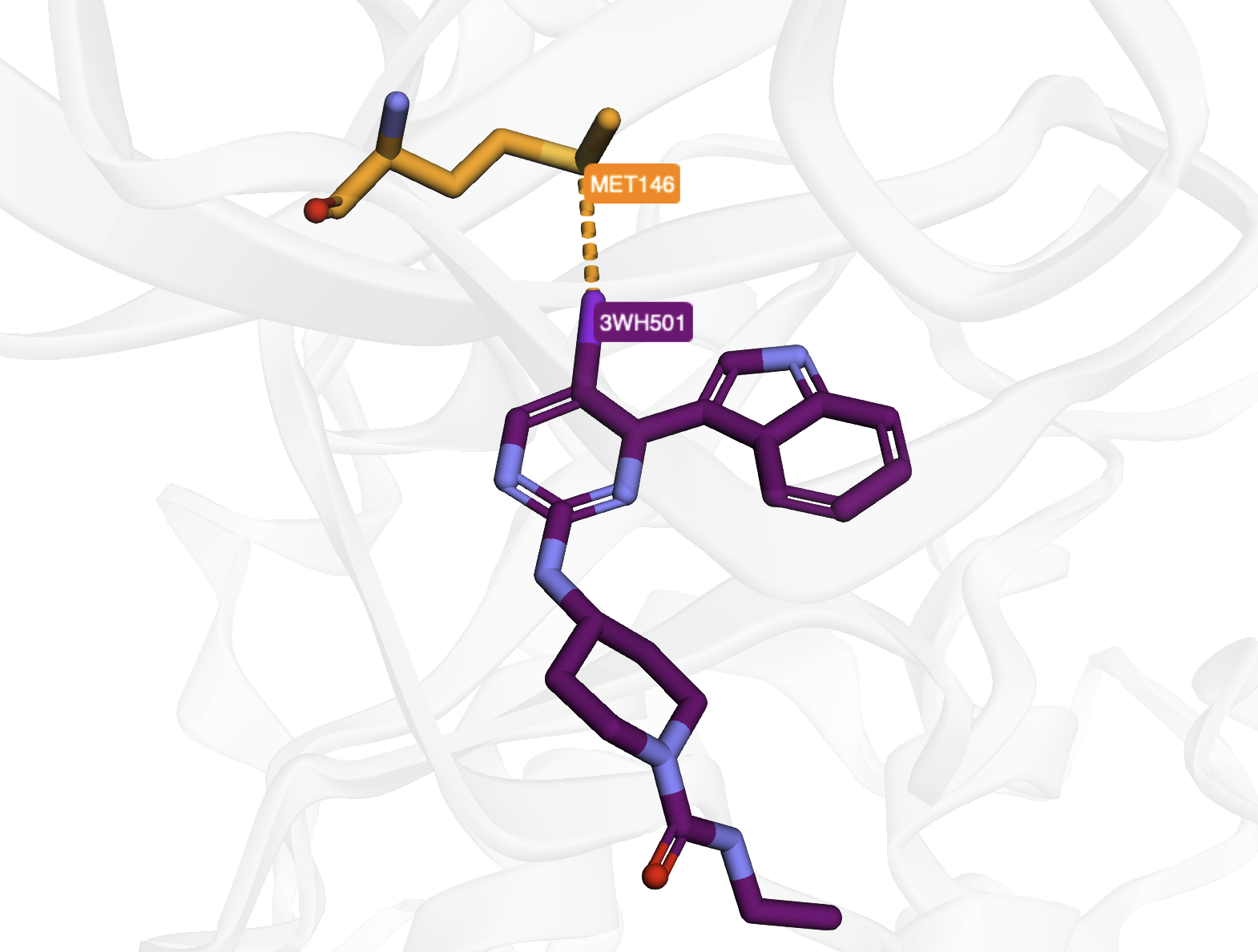
Hydrogen Bonds in PDB Entry 4x21#
Prerequisites#
To run the notebooks locally, install the required dependencies:
pip install hbat py3Dmol pandas jupyter graphviz
Note: The graphviz Python package also requires the Graphviz system software:
Ubuntu/Debian:
sudo apt-get install graphvizmacOS:
brew install graphvizWindows: Download from graphviz.org
Running the Notebooks#
Using Jupyter Notebook#
jupyter notebook
Navigate to the notebooks directory and open the desired notebook.
Using JupyterLab#
jupyter lab
Using VS Code#
Open the notebook file in VS Code
Select the Python kernel
Run cells interactively
Using Google Colab#
Click the “Open in Colab” badge next to any notebook in the table above to run it directly in your browser without any local installation.
Data Files#
The notebooks use example PDB files from the example_pdb_files/ directory:
6rsa.pdb- Ribonuclease A structure (hydrogen bonds, π interactions)4x21.pdb- Crystal structure with halogen bonds